





|
Даю задания для студентов. Они хотят повторно пройти курс. Как отписаться от выполненного курса, что бы пройти его заново? |
Инспектор
Вы можете этот курс.
Опубликован: 29.08.2012 | Доступ: платный | Студентов: 407 / 0 | Длительность: 08:05:00
Специальности: Разработчик интернет-проектов
Практическая работа 7:
Разработка AJAX-приложения
Аннотация: В ходе выполнения данной работы учащиеся разработают AJAX-приложение, используя MSAjaxControlToolkit.
Ключевые слова: AJAX, asynchronous, AND, XML, обмен данными, веб-страница, сайт, инструментарий, архив, пункт, меню, файл, элементы управления
Технология AJAX (Asynchronous Javascript and XML - "асинхронный JavaScript и XML") обеспечивает обмен данными между браузероми веб-сервером в "фоновом" режиме. В результате, при обновлении данных, веб-страница не перезагружается полностью, и веб-приложения становятся более быстрыми и удобными.
Хорошая новость для разработчиков! Microsoft разработала бесплатный инструмент AJAXControlToolkit, который можно скачать по адресу: http://ajaxcontroltoolkit.codeplex.com/. В состав инструментария входит около сорока элементов управления. На этом занятии мы познакомимся с элементами управления Accordionи Slideshow.
Пример 1. Знакомство с элементом управления Accordion
Для начала нам необходимо скачать AJAXControlToolkit. Это можно сделать, зайдя на сайт http://ajaxcontroltoolkit.codeplex.com/. Инструментарий представляет собой архив с именем AjaxControlToolkit.Binary.NET4. Скачанный архив нужно разархивировать. Запустите VisualStudio
и создайте web-сайт с именем AJAX.
После создания нового веб-сайта необходимо добавить AJAXControlToolkit к панели инструментов.
Для этого нужно нажать: View -> Toolbox
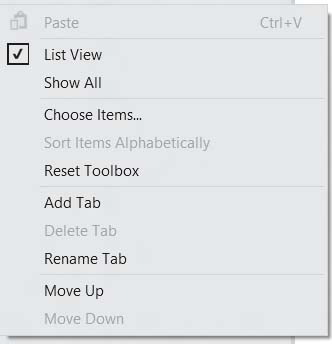
Затем на пустом месте панели инструментов щелкнуть правой кнопкой мыши и выбрать пункт AddTab
Назовите закладку AJAX Control Toolkit
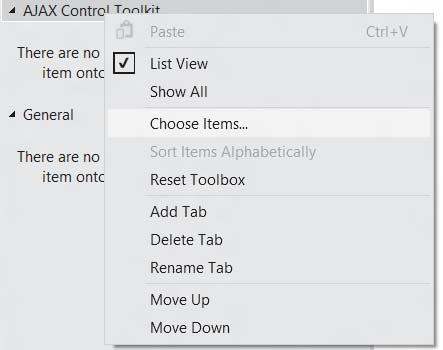
Нажмите по закладке правой кнопкой мыши и выберите пункт ChooseItems…
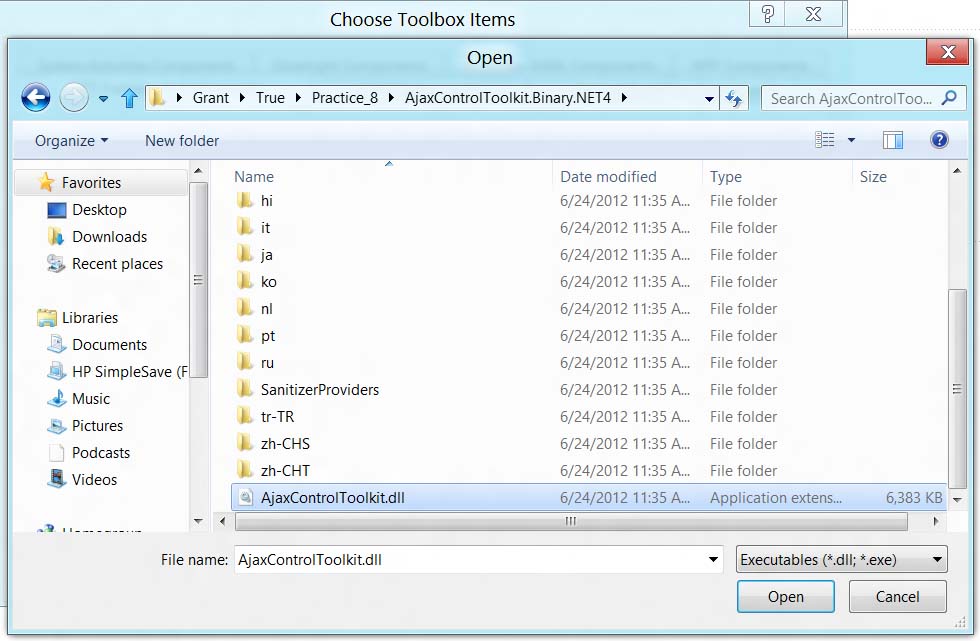
В появившемся меню нажмите кнопку Brows и выберите файл AjaxControlToolkit.dll.
Нажмите кнопку Open. После этого библиотека добавится к нашему проекту
А в закладке AJAXControlToolkit появятся новые элементы управления.
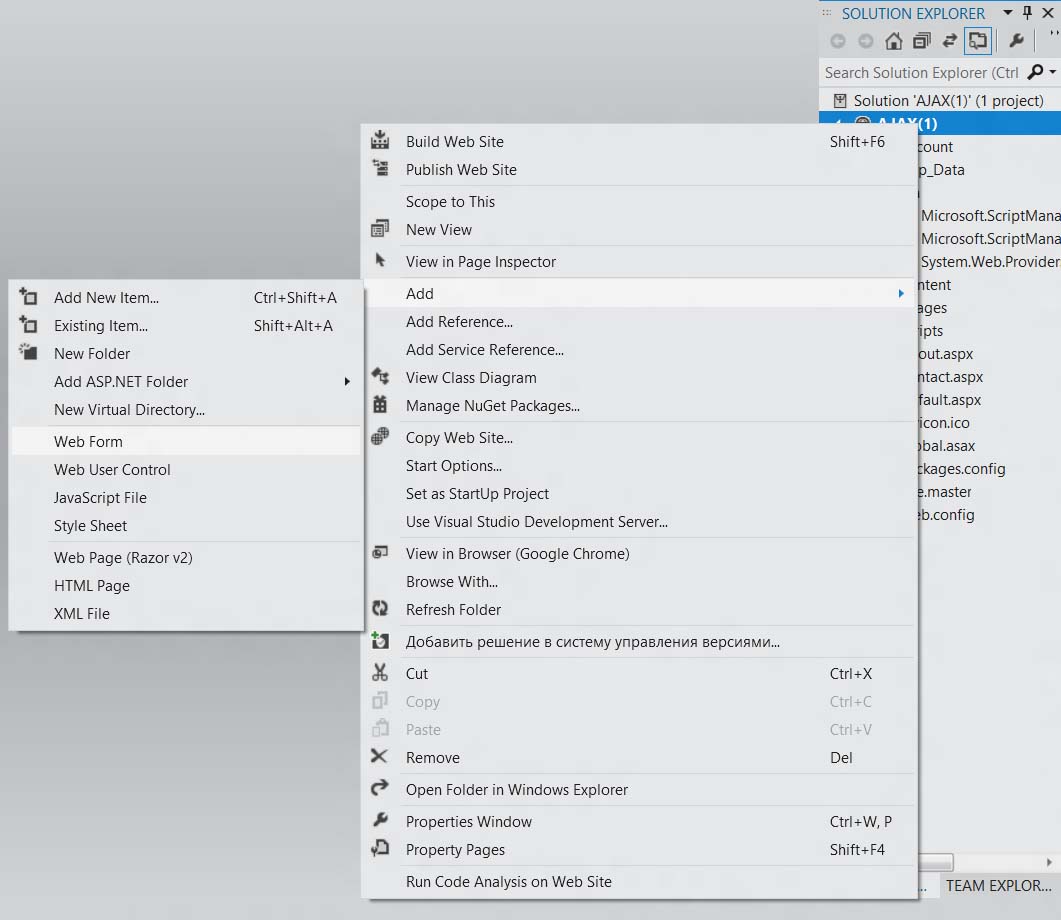
Добавьте web-форму.
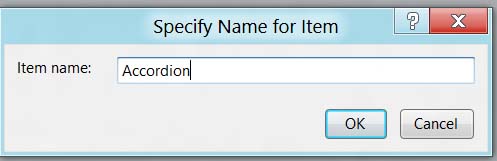
Назовите .aspx-файл именем Accordion
Введите следующий код:
<%@PageLanguage="C#"AutoEventWireup="true"%>
<%@Registerassembly="AjaxControlToolkit"namespace="AjaxControlToolkit"
tagprefix="asp"%>
<!DOCTYPEhtmlPUBLIC"-//W3C//DTD XHTML 1.0 Transitional//EN""
http://www.w3.org/TR/xhtml1/DTD/xhtml1-transitional.dtd">
<htmlxmlns="http://www.w3.org/1999/xhtml">
<headid="Head1"runat="server">
<title>Simple Accordion</title>
<styletype="text/css">
.accordion {
width: auto;
}
.accordionHeader {
border: 1pxsolid#2F4F4F;
color: white;
background-color: #2E4d7B;
font-family: Arial, Sans-Serif;
font-size: 12px;
font-weight: bold;
padding: 5px;
margin-top: 5px;
cursor: pointer;
}
.accordionHeaderSelected {
border: 1pxsolid#2F4F4F;
color: white;
background-color: #5078B3;
font-family: Arial, Sans-Serif;
font-size: 12px;
font-weight: bold;
padding: 5px;
margin-top: 5px;
cursor: pointer;
}
.accordionContent {
background-color: #D3DEEF;
border: 1pxdashed#2F4F4F;
font-family: 'Comic Sans MS';
border-top: none;
padding: 5px;
padding-top: 10px;
}
h1 {
font-family: 'Comic Sans MS'; font-size: larger;color: #2E4d7B
}
h3 {
font-family: 'Comic Sans MS'; font-size: medium;color: #2E4d7B
}
</style>
</head>
<body>
<h1>Словарь терминов по биоинформатике</h1>
<h3>По материалам сайта Википедия</h3>
<formid="form1"runat="server">
<div>
<asp:ToolkitScriptManagerID="ToolkitScriptManager1"runat="server">
</asp:ToolkitScriptManager>
<asp:Accordion
ID="Accordion1"
CssClass="accordion"
HeaderCssClass="accordionHeader"
HeaderSelectedCssClass="accordionHeaderSelected"
ContentCssClass="accordionContent"
runat="server">
<Panes>
<asp:AccordionPaneID="AccordionPane1"runat="server">
<Header>Анализ генетических последовательностей</Header>
<Content>
C тех пор как в 1977 году был секвенирован фаг Phi-X174, последовательности ДНК всё большего числа организмов
были дешифрованы и сохранены в базах данных. Эти данные используются для определения последовательностей
белков и регуляторных участков. Сравнение генов в рамках одного или разных видов может продемонстрировать
сходство функций белков или отношения между видами (таким образом могут быть составлены Филогенетические деревья).
С возрастанием количества данных уже давно стало невозможным вручную анализировать последовательности.
В наши дни для поиска по геномам тысяч организмов, состоящих из миллиардов пар нуклеотидов
используются компьютерные программы. Программы могут однозначно сопоставить (выровнять)
похожие последовательности ДНК в геномах разных видов; часто такие последовательности несут сходные функции,
а различия возникают в результате мелких мутаций, таких как замены отдельных нуклеотидов,
вставки нуклеотидов, и их "выпадения" (делеции).
Один из вариантов такого выравнивания применяется при самом процессе секвенирования.
Так называемая техника "дробного секвенирования" (которая была, например,
использована Институтом Генетических Исследований для секвенирования первого бактериального генома,
Haemophilusinfluenzae) вместо полной последовательности нуклеотидов даёт последовательности коротких фрагментов ДНК
(каждый длиной около 600-800 нуклеотидов). Концы фрагментов накладываются друг на друга и,
совмещённые должным образом, дают полный геном. Такой метод быстро даёт результаты секвенирования,
но сборка фрагментов может быть довольно сложной задачей для больших геномов.
В проекте по расшифроке генома человека сборка заняла несколько месяцев компьютерного времени.
Сейчас этот метод применяется для практически всех геномов,
и алгоритмы сборки геномов являются одной из острейших проблем биоинформатики на сегодняшний момент.
<br/>Другим примером применения компьютерного анализа последовательностей является
автоматический поиск генов и регуляторных последовательностей в геноме.
Не все нуклеотиды в геноме используются для задания последовательностей белков.
Например, в геномах высших организмов, большие сегменты ДНК явно не кодируют белки и их функциональная роль неизвестна.
Разработка алгоритмов выявления кодирующих белки участков генома является важной задачей современной биоинформатики.
<br/>Биоинформатика помогает связать геномные и протеомные проекты, к примеру,
помогая в использовании последовательности ДНК для идентификации белков.
</Content>
</asp:AccordionPane>
<asp:AccordionPaneID="AccordionPane2"runat="server">
<Header>Аннотациягеномов</Header>
<Content>
В контексте геномики аннотация - процесс маркировки генов и других объектов в последовательности ДНК.
Первая программная система аннотации геномов была создана в 1995 году Оуэном Уайтом (англ. OwenWhite),
работавшим в команде, секвенировавшей и проанализировавшей первый декодированный геном свободноживущего организма,
бактерии Haemophilusinfluenzae. Доктор Уайт построил систему для нахождения генов,
тРНК и других объектов ДНК и сделал первые обозначения функций этих генов.
Большинство современных систем работают сходным образом, но эти программы постоянно развиваются и улучшаются.
</Content>
</asp:AccordionPane>
<asp:AccordionPaneID="AccordionPane3"runat="server">
<Header>Вычислительная эволюционная биология</Header>
<Content>
Эволюционная биология исследует происхождение и появление видов, также как их развитие с течением времени.
Информатика помогает эволюционным биологам в нескольких аспектах:
<br/>изучать эволюцию большого числа организмов, измеряя изменения в их ДНК, а не только в строении или физиологии;
<br/>сравнивать целые геномы (см. BLAST), что позволяет изучать более комплексные эволюционные события, такие как:
дупликация генов, латеральный перенос генов, и предсказывать бактериальные специализирующие факторы;
<br/>строить компьютерные модели популяций, чтобы предсказать поведение системы во времени;
<br/>отслеживать появление публикаций, содержащих информацию о большом количестве видов.
<br/>Область в компьютерных науках, которая использует генетические алгоритмы,
часто путают с компьютерной эволюционной биологией. Работа в этой области использует специализированное
программное обеспечение для улучшения алгоритмов и вычислений и основывается на эволюционных принципах,
таких, как репликация, диферсификация через рекомбинацию или мутации, и выживании в естественном отборе.
</Content>
</asp:AccordionPane>
<asp:AccordionPaneID="AccordionPane4"runat="server">
<Header>Оценка биологического разнообразия</Header>
<Content>
Биологическое разнообразие экосистемы может быть определено как полная генетическая совокупность определённой среды,
состоящая из всех обитающих видов, была бы это биоплёнка в заброшенной шахте, капля морской воды, горсть земли или вся
биосфера планеты Земля. Для сбора видовых имён, описаний, ареала распространения, генетической информации
используются базы данных. Специализированное программное обеспечение применяется для поиска, визуализации и
анализа информации, и, что более важно, предоставления её другим людям. Компьютерные симуляторы моделируют такие вещи,
как популяционная динамика, или вычисляют общее генетическое здоровье культуры в агрономии.
Один из важнейших потенциалов этой области заключается в анализе последовательностей ДНК или полных геномов
целых вымирающих видов, позволяя запомнить результаты генетического эксперимента природы в компьютере и
возможно использовать вновь в будущем, даже если эти виды полностью вымрут.
<br/>Часто из области рассмотрения биоинформатики выпадают методы оценки других компонентов биоразнообразия -
таксонов (в первую очередь видов) и экосистем. В настоящее время математические основания биоинформационных методов
для таксонов представлены в рамках такого научного направления как фенетика, или численная таксономия.
Методы анализа структуры экосистем рассматриваются специалистами таких направлений как системная экология, биоценометрия.
</Content>
</asp:AccordionPane>
</Panes>
</asp:Accordion>
</div>
</form>
</body>
</html>
Александр Лобанов